Deep Learning for Computational Biology
Review:
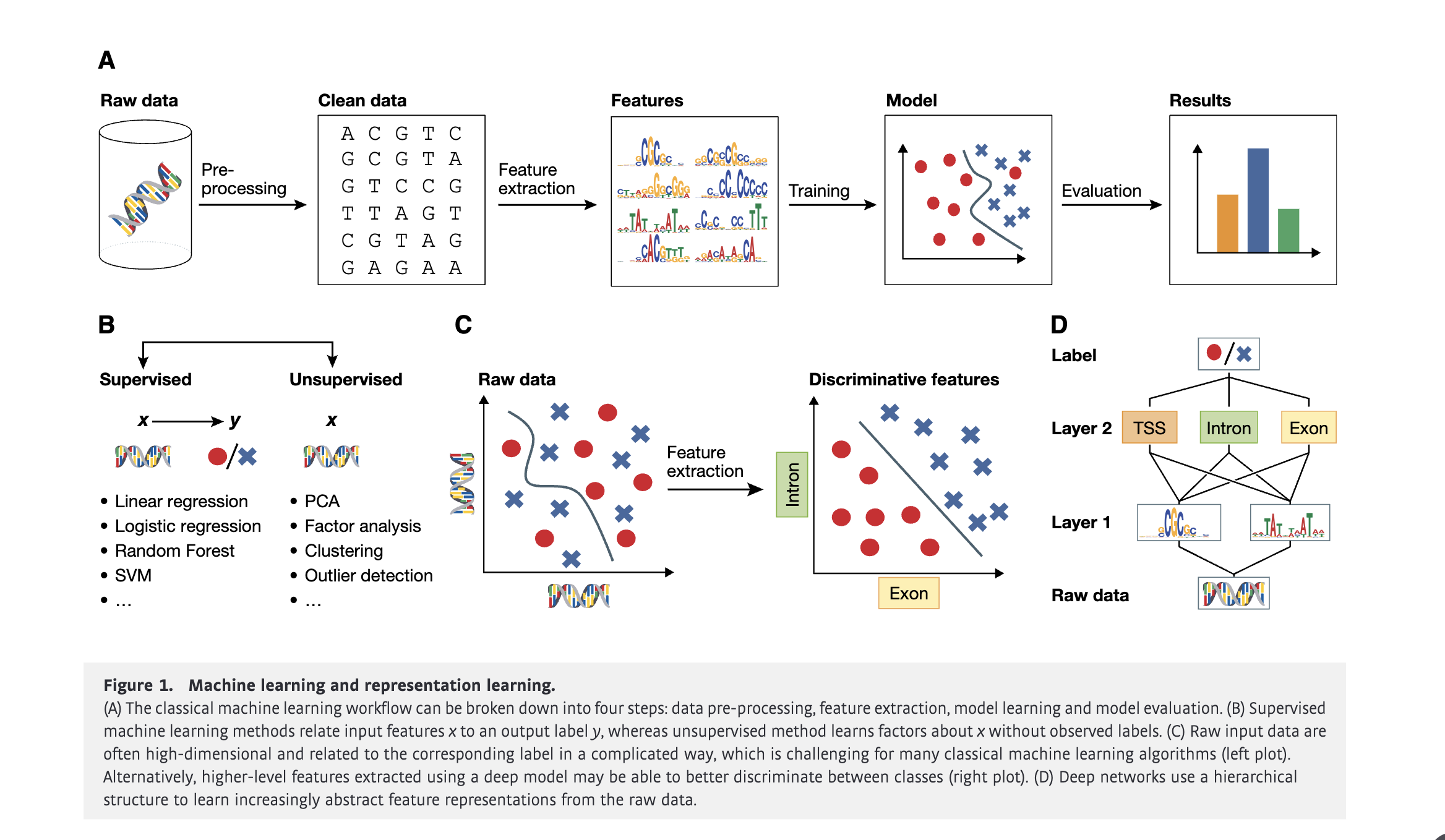
Technological advances in genomics and imaging have led to an explosion of molecular and cellular profiling data from large numbers of samples. This rapid increase in biological data dimension and acquisition rate is challenging conventional analysis strategies. Modern machine learning methods, such as deep learning, promise to leverage very large data sets for finding hidden structure within them, and for making accurate predictions.
In this review, we discuss applications of this new breed of analysis approaches in regulatory genomics and cellular imaging. We provide background of what deep learning is, and the settings in which it can be successfully applied to derive biological insights. In addition to presenting specific applications and providing tips for practical use, we also highlight possible pitfalls and limitations to guide computational biologists when and how to make the most use of this new technology.
Show more
Convolutional Neural Networks in Pharmacogenomics
Review:
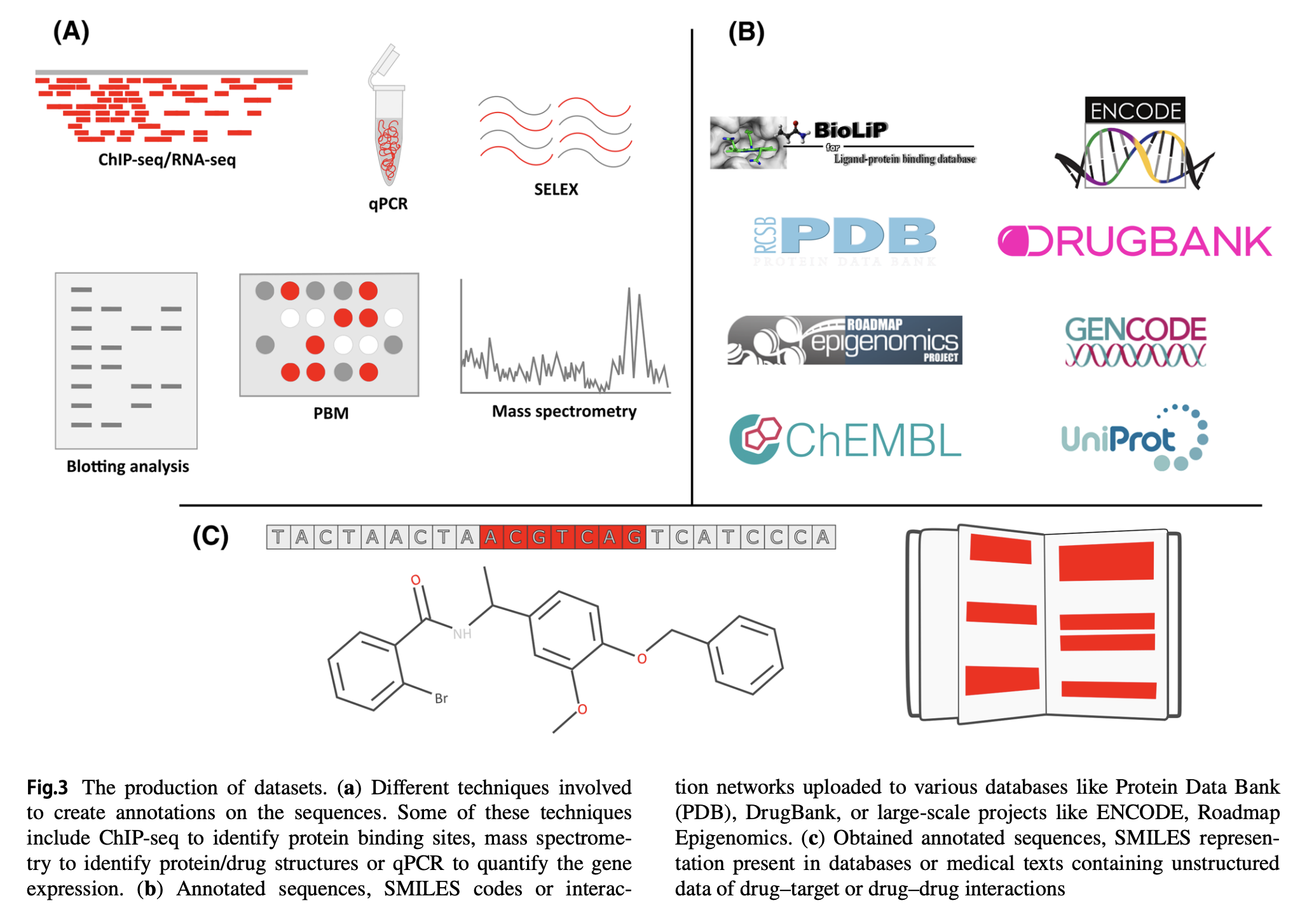
Convolutional neural networks (CNNs) have been used to extract information from various datasets of diferent dimensions. This approach has led to accurate interpretations in several subfelds of biological research, like pharmacogenomics, addressing issues previously faced by other computational methods. With the rising attention for personalized and precision medicine, scientists and clinicians have now turned to artifcial intelligence systems to provide them with solutions for therapeutics development.
CNNs have already provided valuable insights into biological data transformation. Due to the rise of interest in precision and personalized medicine, in this review, we have provided a brief overview of the possibilities of implementing CNNs as an efective tool for analyzing one-dimensional biological data, such as nucleotide and protein sequences, as well as small molecular data, e.g., simplifed molecular-input line-entry specifcation, InChI, binary fngerprints, etc., to categorize the models based on their objective and also highlight various challenges. The review is organized into specifc research domains that participate in pharmacogenomics for a more comprehensive understanding. Furthermore, the future intentions of deep learning are outlined.
Show more
Supervised Machine Learning To Predict Solvation Gibbs Energy
Research:
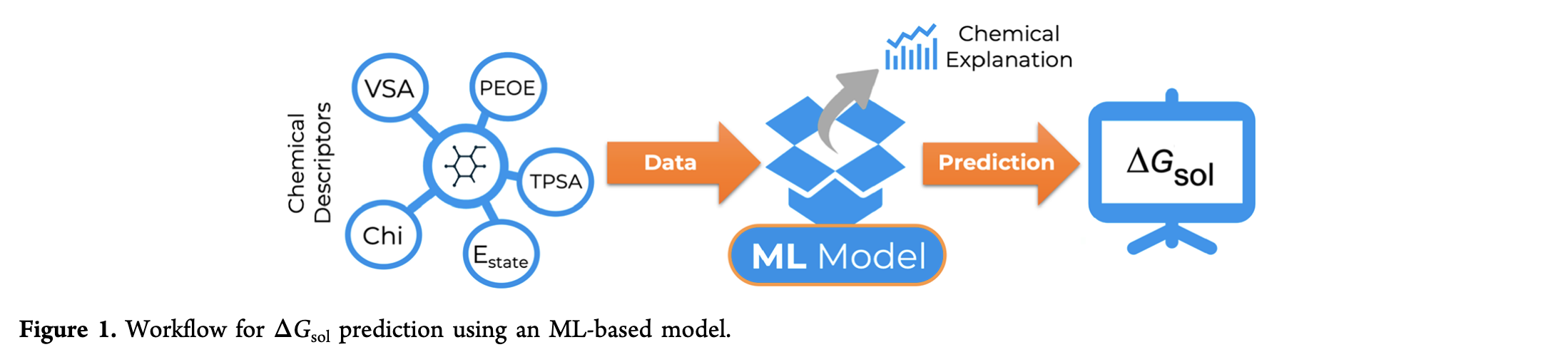
Many challenges persist in developing accurate computational models for predicting solvation free energy (ΔGsol).Despite recent developments in Machine Learning (ML) methodologies that outperformed traditional quantum mechanical models,several issues remain concerning explanatory insights for broad chemical predictions with an acceptable speed−accuracy trade-off.To overcome this, we present a novel supervised ML model to predict the ΔGsol for an array of solvent−solute pairs.
Using twodifferent ensemble regressor algorithms, we made fast and accurate property predictions using open-source chemical features,encoding complex electronic, structural, and surface area descriptors for every solvent and solute. By integrating molecular propertiesand chemical interaction features, we have analyzed individual descriptor importance and optimized our model though explanatoryinformation form feature groups. On aqueous and organic solvent databases, ML models revealed the predictive relevance of soluteswith increasing polar surface area and decreasing polarizability, yielding better results than state-of-the-art benchmark NeuralNetwork methods (without complex quantum mechanical or molecular dynamic simulations). Both algorithms successfullyoutperformed previous ΔGsol predictions methods, with a maximum absolute error of 0.22 ± 0.02 kcal mol−1, further validated in anexternal benchmark database and with solvent hold-out tests. With these explanatory and statistical insights, they allow a thoughtfulapplication of this method for predicting other thermodynamic properties, stressing the relevance of ML modeling for furthercomplex computational chemistry problems.
Show more
Graph Respresentations for Functional Residue Identification
Research:
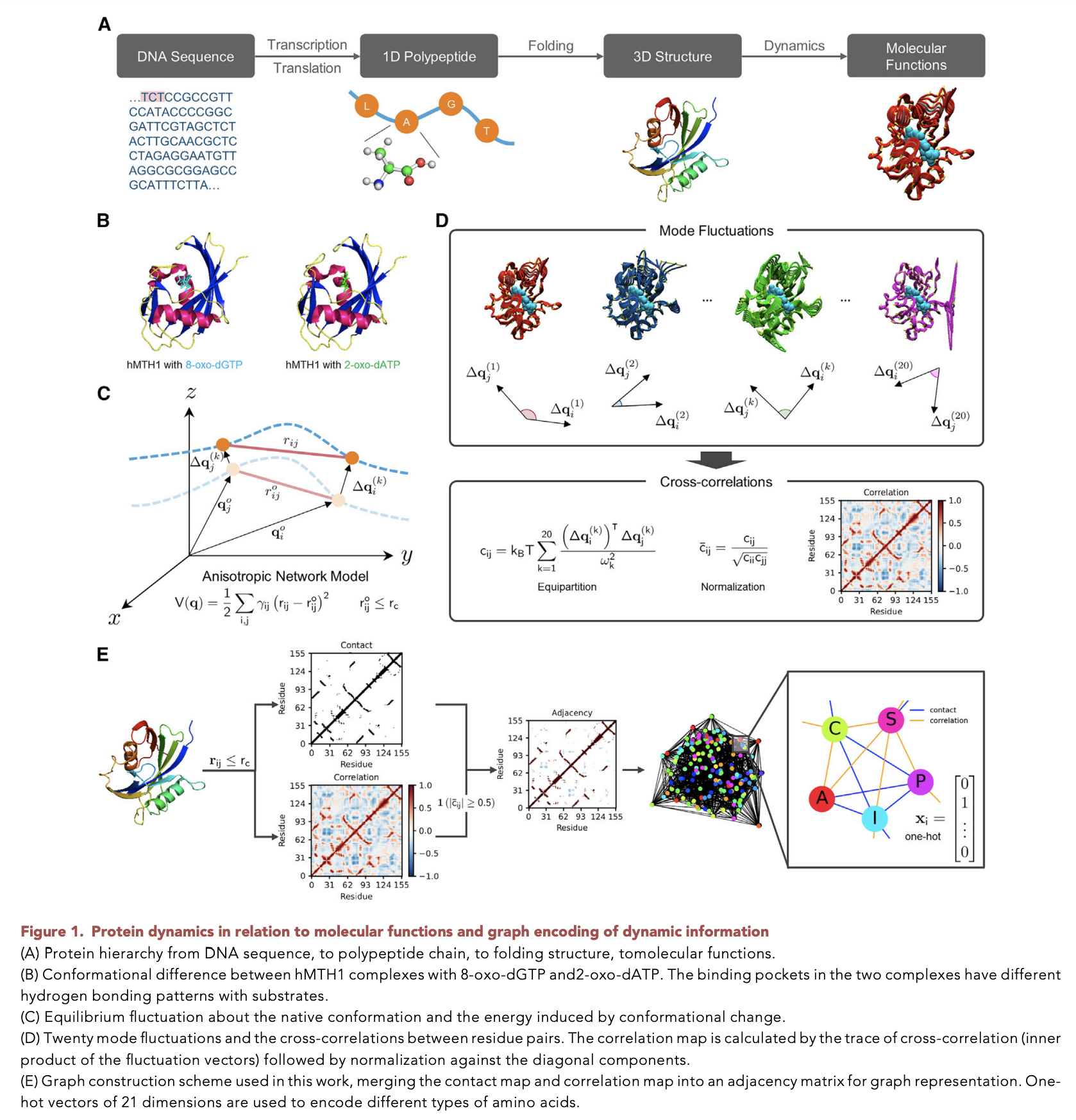
Recent advances in protein function prediction exploit graph-based deep learning approaches to correlate the structural and topological features of proteins with their molecular functions. However, proteins in vivo are not static but dynamic molecules that alter conformation for functional purposes. Here, we apply normal mode analysis to native protein conformations and augment protein graphs by connecting edges between dynamically correlated residue pairs.
In the multilabel function classification task, our method demonstrates a remarkable performance gain based on this dynamics-informed representation. The proposed graph neural network, ProDAR, increases the interpretability and generalizability of residue-level annotations and robustly reflects structural nuance in proteins. We elucidate the importance of dynamic information in graph representation by comparing class activation maps for hMTH1, nitrophorin, and SARS-CoV-2 receptor binding domain. Our model successfully learns the dynamic fingerprints of proteins and pinpoints the residues of functional impacts, with vast untapped potential for broad biotechnology and pharmaceutical applications.
Show more
Understanding Structural Variability Using Protein Networks
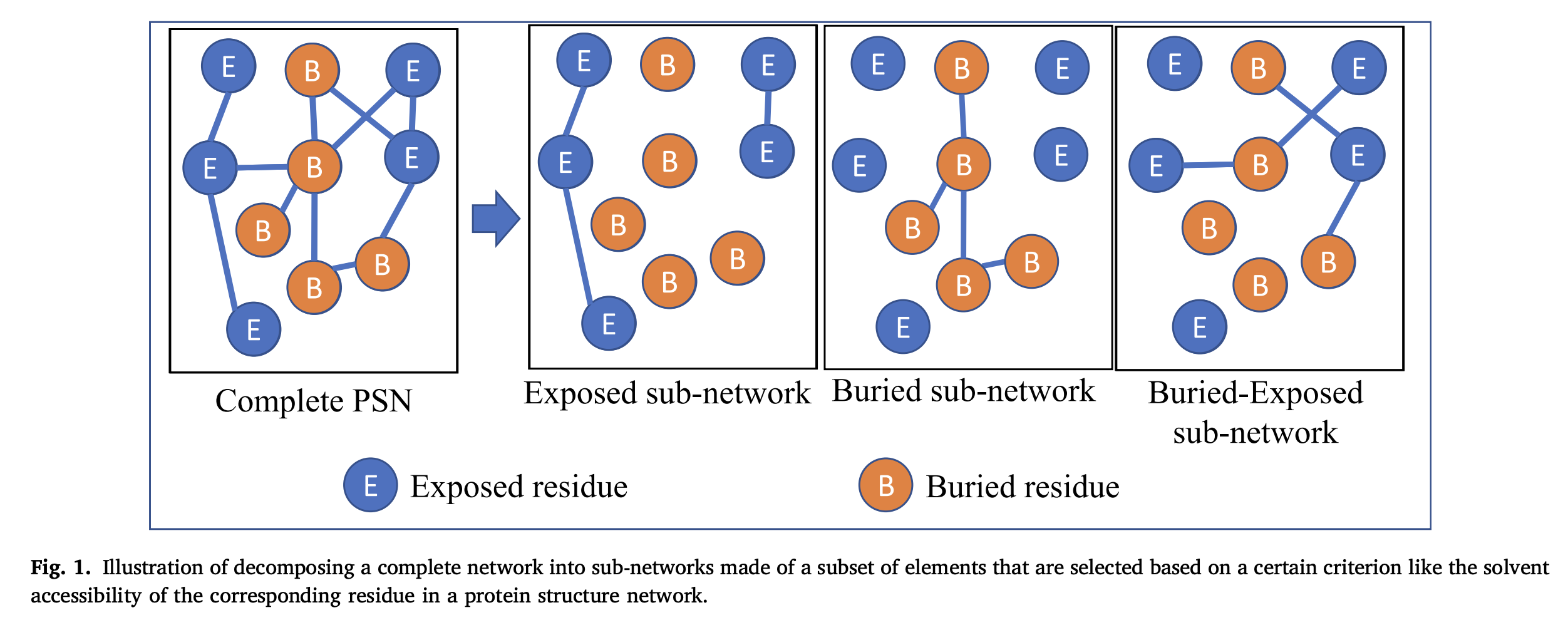
Research:
Proteins perform their function by accessing a suitable conformer from the ensemble of available conformations. The conformational diversity of a chosen protein structure can be obtained by experimental methods under different conditions. A key issue is the accurate comparison of different conformations. A gold standard used for such a comparison is the root mean square deviation (RMSD) between the two structures.
While extensive refinements of RMSD evaluation at the backbone level are available, a comprehensive framework including the side chain interaction is not well understood. Here we employ protein structure network (PSN) formalism, with the non-covalent interactions of side chain, explicitly treated. The PSNs thus constructed are compared through graph spectral method, which provides a comparison at the local and at the global structural level. In this work, PSNs of multiple crystal conformers of single-chain, single-domain proteins, are subject to pair-wise analysis to examine the dissimilarity in their network topologies and in order to determine the conformational diversity of their native structures. This information is utilized to classify the structural domains of proteins into different categories. It is observed that proteins typically tend to retain structure and interactions at the backbone level. However, some of them also depict variability in either their overall structure or only in their inter-residue connectivity at the sidechain level, or both. Variability of sub-networks based on solvent accessibility and secondary structure is studied. The types of specific interactions are found to contribute differently to structure variability. An ensemble analysis by computing the mathematical variance of edge-weights across multiple conformers provided information on the contribution to overall variability from each edge of the PSN. Interactions that are highly variable are identified and their impact on structure variability has been discussed with the help of a case study. The classification based on the present side-chain network-based studies provides a framework to correlate the structure-function relationships in protein structures.
Show more
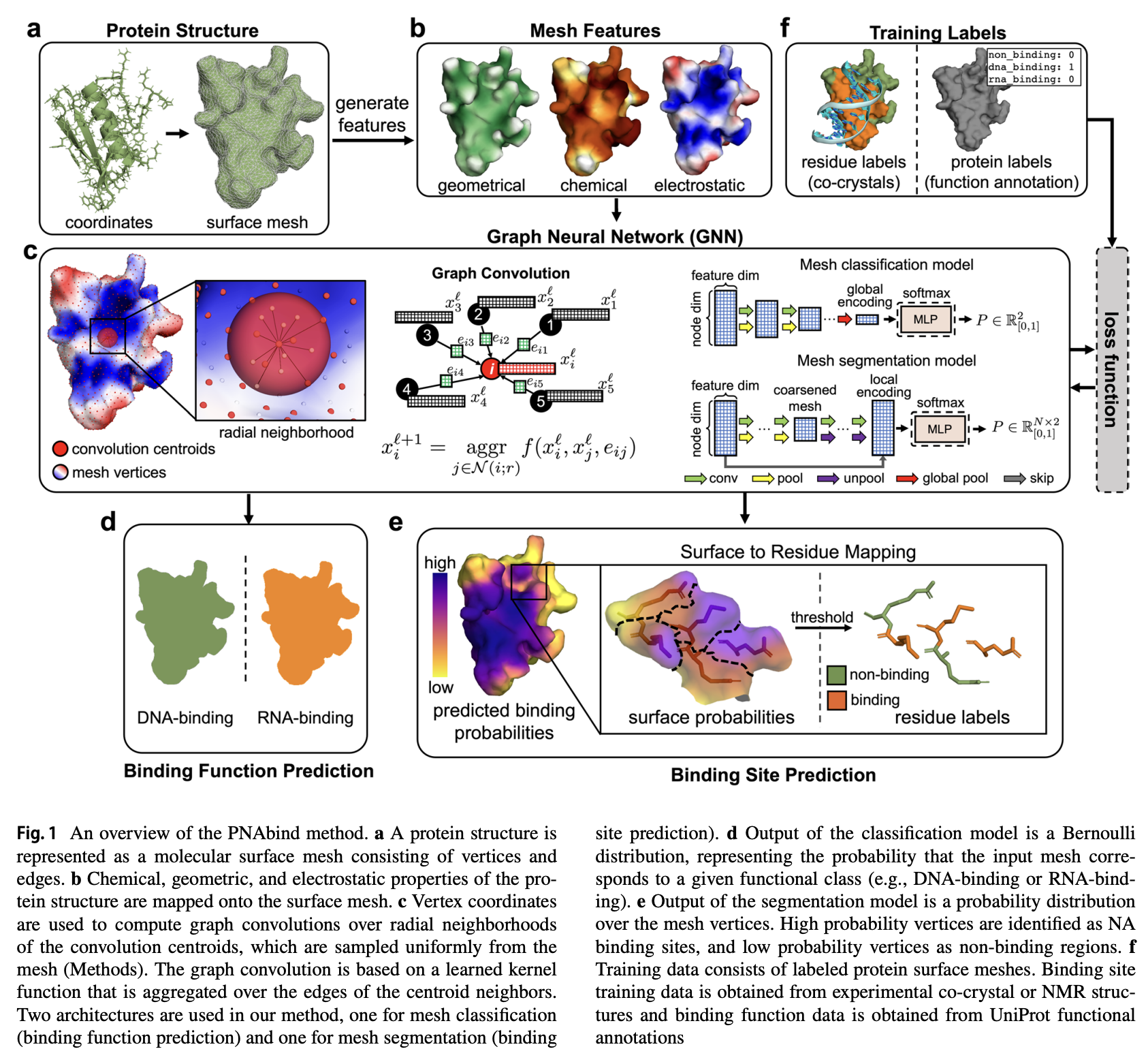
Review:
Protein-nucleic acid (PNA) binding plays critical roles in the transcription, translation, regulation, and three-dimensional organization of the genome. Structural models of proteins bound to nucleic acids (NA) provide insights into the chemical, electrostatic, and geometric properties of the protein structure that give rise to NA binding but are scarce relative to models of unbound proteins. We developed a deep learning approach for predicting PNA binding given the unbound structure of a protein that we call PNAbind.
Our method utilizes graph neural networks to encode the spatial distribution of physicochemical and geometric properties of protein structures that are predictive of NA binding. Using global physicochemical encodings, our models predict the overall binding function of a protein, and using local encodings, they predict the location of individual NA binding residues. Our models can discriminate between specifcity for DNA or RNA binding, and we show that predictions made on computationally derived protein structures can be used to gain mechanistic understanding of chemical and structural features that determine NA recognition. Binding site predictions were validated against benchmark datasets, achieving AUROC scores in the range of 0.92–0.95. We applied our models to the HIV-1 restriction factor APOBEC3G and showed that our model predictions are consistent with and help explain experimental RNA binding data.
Show more